Chemical Dynamics – Watching Molecules in Time and Space
Peter M. Weber
Chemists have become immensely adept in figuring out the behavior of molecules. And Chemistry works! After all, who could argue with Chemistry’s importance to synthesizing drug molecules to treat illnesses, designing materials with highly specific properties, or creating batteries for energy storage that keep our laptops going and – looking forward – our cars running? There is no doubt: innovations in Chemistry have driven our society forward.
Yet all these successes put into sharp contrast one thing that Chemists are still struggling with: we have not observed real molecules while they are actually doing their chemistry! We have not been able, yet, to watch a molecule transform itself from one structure to another. But we are close – very close. In the Weber group, we are pioneering new technologies that are just now allowing us to realize this dream.
What does it take? We all know that molecules are small, on the order of Ångstroms, i.e. 10-10 m. In addition, they are fast: molecular vibrations happen on a time scale of femtoseconds (10-15 s). So, to watch molecules do their magical Chemistry, we need tools that allow us to see Ångstrom distances on femtosecond time scales. This is not easy.
Two techniques feature prominently in the Weber group’s approach to watch molecules do their dance. One is spectroscopic and the other uses diffraction. Both work on the ultrafast time scales required to time-resolve the molecular motions.
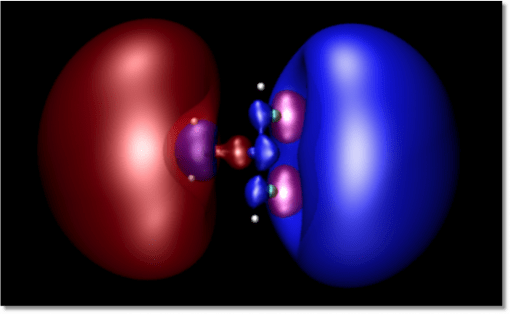
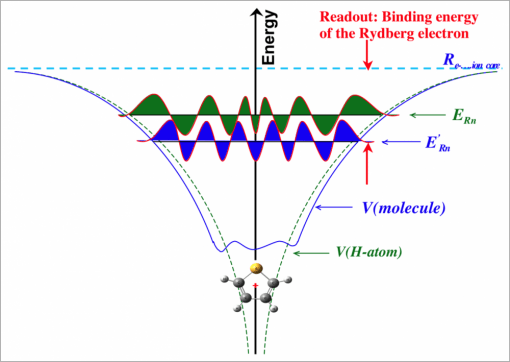
The spectroscopic method is based on time-resolved multi-photon ionization photoelectron spectroscopy. It takes advantage of the fact, which we have discovered in our recent research, that the energy with which a Rydberg electron is bound to a positive ion core is very sensitive to the molecular structure (see Figures 1 and 2). In fact, recent research results encourage us to think that we can, in fact, determine the molecular structure from the spectrum. But that is still a bit out there. For now we use it to fingerprint the molecular structure.
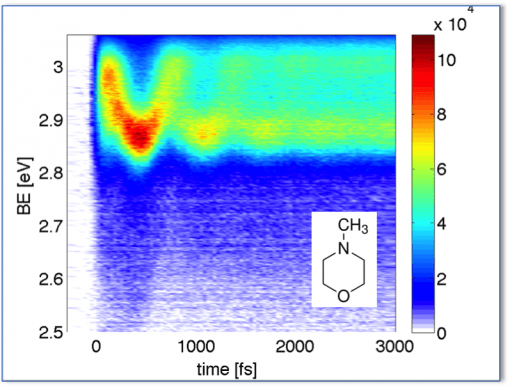
To watch molecules doing Chemistry, we induce the chemical reaction with a femtosecond laser pulse, and we watch the molecular reaction by following the time evolution of the Rydberg electron binding energy. We have applied this to numerous model systems and for details please check out the following references: 39, 42, 45, 48, 50, 54, 55, 56, 57, 58, 60, 63, 65, 67, 69, 72, 74, 76. Figure 3 shows the structural oscillations induced in a molecule upon excitation with a short ultraviolet pulse.
The diffraction method takes a very different tack. Chemists use diffraction, mostly x-ray diffraction, to determine structures of molecules large and small. But those are typically frozen structures, of molecules in crystals. Chemical reactions don’t just happen in crystalline phases, with all molecules in their ground state structure. So, how could one turn diffraction into a time-resolved technique?
The answer is to generate ultrashort pulses. Either of x-ray, for x-ray diffraction, or of electrons, to do electron diffraction. Both work in principle. In practice, electrons traditionally had the edge on gas phase systems since electron scattering cross sections are much, much larger than x-ray cross sections. So, when we first explored this area we have used time-resolved electron diffraction, see publications 18, 19, 20, 23, 28, 33, 36, 38, 40, 41, 43, 46, 51, 52, 75.
But the advent of free electron lasers has changed the equation: even though x-ray scattering cross sections are so small, one can still get good diffraction if one just has enough x-rays in a pulse. And it is now possible to get enough x-rays in an ultrashort pulse, namely from free electron lasers. We do our experiments at the Linac Coherent Light Source (LCLS) free electron laser at the SLAC National Accelerator Laboratory. Our experimental setup is at the end of a kilometer-long electron accelerator that generates coherent, ultrashort x-ray pulses with durations short enough to watch molecular motions.
To explore chemical dynamics with the LCLS light source, we resort again to pump-probe experiments: a laser pulse excites a reaction, and the x-ray pulse from LCLS probes the progress of the reaction. To record the molecular movie we take snapshots of the x-ray diffraction patterns as a function of the time delay between the optical pump and the x-ray probe pulses. There is one paper coming out shortly (Faraday Discuss., 2014, 171, 1–11), and several others are in the works and will be posted here soon.
All our experimental studies are deeply embedded in collaborations with leading theorists. For example, we have simulated how diffraction patterns of molecules in excited states should be expected to look. And we have teamed up with Hannes Jónsson, Brown University and University of Iceland, to expand the applicability of DFT to Rydberg states. Not only do these interactions with theorists deepen our understanding of the molecular processes, they also allow us to ever advance our arsenal of experimental tools. For papers on theory please see references 23, 28, 33, 38, 40, 65, 77.